Аміотрофія — хвороба, описана вченими в кінці 19 століття і охарактеризована змінами певних груп м’язів, нервів на периферії і самого спинного мозку. У наукових працях було помічена атрофическая симетричність клітин передніх корінців і рогів спинного мозку. Пізніше виділили форму захворювання легшу, де уражаються тільки клітини передніх рогів спинного мозку, назвали її — нозологічної. Спинальная м’язова атрофія — спадкове генетичне захворювання. Відбувається втрата м’язової активності за рахунок деградації нейронів передніх рогів спинного мозку. Страждає поперечнополосата мускулатура нижнього пояса, шиї і голови, верхній плечовий пояс задіяний в меншій мірі. У людини спостерігаються порушення мимовільних рухів, наприклад, повзання у дитини або ходьби. Але психічно людина не страждає, залишається чутливість при спінальної атрофії. В чому причина? Захворювання розвивається досить рідко, так як передається по аутосомно-рецесивним типом. Це означає, що у обох батьків має бути зміна генетичного матеріалу в п’ятій хромосомі (SMN), тоді швидше за все (але не 100%), народитися дитина з таким захворюванням. В процесі цього збою відбувається зменшення вироблення білка SMN, який і веде до втрати моторних нейронів.
Класифікація
 Є класифікація даного захворювання. Розподіл відбувається за віковими характеристиками, складності протікання і вікових особливостей:
Є класифікація даного захворювання. Розподіл відбувається за віковими характеристиками, складності протікання і вікових особливостей:
- Спинальная м’язова атрофія 1 типу або дитячий (сма 1), так само можна зустріти назву: хвороба Вердінга- Гоффмана. Страждають діти до першого півроку життя. Часто результат смертельний. У новонароджених виникають стійкі порушення смоктального рефлексу, а також ковтання.
- Атрофія 2 типу або проміжний (сма 2) або хвороба Дубовиця. Хворіють діти від 7 місяців до 1 року. При цій формі діти можуть харчуватися і ковтати, але самостійно ходити або сидіти не можуть. На жаль, гинуть діти через супутніх захворювань, наприклад, від застійних пневмоній через малорухомості.
- 3 тип або юнацький (ювенільний) (сма) 3 або хвороба Кюгельберга — Веландера. Захворювання починається з 15 років і може бути навіть у дорослих. Такі пацієнти можуть самостійно стояти, але пересуваються в інвалідному кріслі.
- 4 тип або дорослий — (сма 4) або доросла форма. Виявляється з 35 років, дуже повільно розвивається, але може привести до повної втрати рухової активності.
Клінічна картина
Захворювання ділитися на 4 типи. Тяжкість хвороби і її результат залежить від складності перебігу захворювання від віку пацієнта. Як правило, в будь-якій формі ставлять інвалідність. У важких типах хвороба може вимагати постійної медичної помощі.В При будь-якому з видів СМА не страждає чутливість, так як в процесі не беруть участь чутливі нервові волокна. Інтелектуальна сторона так само не задіяна, тому дитина легко навчаємо на рівні зі своїми однолітками. А ось серцева і дихальна системи беруть на себе весь удар наслідків цієї хвороби. Смерть настає в основному через затяжні застійних пневмоній або бронхітів, в слідстві практично відсутньої рухової активності.
Тип 1. Хвороба Верднига-Гоффмана
 Спінальної-м’язова атрофія Верднига-Гоффмана починає проявлятися ще у внутрішньоутробному розвитку плода. Захворювання отримало назву за прізвищами вчених, Вердніга і Гофманом були описані одні з перших морфологічних ознак хвороби. Приблизно з двадцять восьмого тижня вагітності спостерігається слабка активність плода. Після народження у дитини починають наростати певні ознаки цього захворювання в перші пів року життя. Дитина постійно лежить, практично знерухомлені, не перевертається, спостерігається тик уражених м’язів, ніжки не згинає. В результаті розвитку хвороби 1 типу відбувається атрофія м’язів, вони значно зменшуються. Глотка, діафрагма, міжреберні і грудні м’язи не забезпечується нервовими клітинами, що ускладнює процес ковтання. Це все веде до застійних пневмоній, їх обов’язково потрібно своєчасно діагностувати, в іншому ж випадку це призведе до летальності. Ще одна характерна риса при захворюванні Верднига Гоффмана — деформація скелета, через ослаблення м’язів, які не здатні утримувати скелет. Якщо дитина намагається сидіти, то при захворюванні Вердніга, як правило, формується сколіоз. Ущільнюється так само грудна клітка, це веде до утруднення дихання і порушення функцій серцево — судинної системи.
Спінальної-м’язова атрофія Верднига-Гоффмана починає проявлятися ще у внутрішньоутробному розвитку плода. Захворювання отримало назву за прізвищами вчених, Вердніга і Гофманом були описані одні з перших морфологічних ознак хвороби. Приблизно з двадцять восьмого тижня вагітності спостерігається слабка активність плода. Після народження у дитини починають наростати певні ознаки цього захворювання в перші пів року життя. Дитина постійно лежить, практично знерухомлені, не перевертається, спостерігається тик уражених м’язів, ніжки не згинає. В результаті розвитку хвороби 1 типу відбувається атрофія м’язів, вони значно зменшуються. Глотка, діафрагма, міжреберні і грудні м’язи не забезпечується нервовими клітинами, що ускладнює процес ковтання. Це все веде до застійних пневмоній, їх обов’язково потрібно своєчасно діагностувати, в іншому ж випадку це призведе до летальності. Ще одна характерна риса при захворюванні Верднига Гоффмана — деформація скелета, через ослаблення м’язів, які не здатні утримувати скелет. Якщо дитина намагається сидіти, то при захворюванні Вердніга, як правило, формується сколіоз. Ущільнюється так само грудна клітка, це веде до утруднення дихання і порушення функцій серцево — судинної системи.
Тип 2. Дитяча форма
 Цей тип захворювання діагностується пізніше, приблизно з півтора року, як тільки дитина робить спроби самостійно повзати або сидіти. Симптоми захворювання у дітей помічають вже при народженні. Перші ознаки — це затримка фізичного розвитку, дуже млява рухова активність, сухожильні рефлекси знижені. Ця форма захворювання переноситься легше, ніж тип Вердніга. Атрофія м’язів протікати повільно, і дитина може проживати до 18 років. Пацієнт може самостійно обслуговувати себе надалі, так само сидіти, стояти, є, але пересуватися тільки за допомогою інвалідного крісла. За рахунок малої рухової активності, можуть приєднаються різні респіраторні інфекції, а також запалення легенів, в найважчих формах їх проявів.
Цей тип захворювання діагностується пізніше, приблизно з півтора року, як тільки дитина робить спроби самостійно повзати або сидіти. Симптоми захворювання у дітей помічають вже при народженні. Перші ознаки — це затримка фізичного розвитку, дуже млява рухова активність, сухожильні рефлекси знижені. Ця форма захворювання переноситься легше, ніж тип Вердніга. Атрофія м’язів протікати повільно, і дитина може проживати до 18 років. Пацієнт може самостійно обслуговувати себе надалі, так само сидіти, стояти, є, але пересуватися тільки за допомогою інвалідного крісла. За рахунок малої рухової активності, можуть приєднаються різні респіраторні інфекції, а також запалення легенів, в найважчих формах їх проявів.
Тип 3. Ювенільний або хвороба Куленберга — Веландера
Характеризується розвитком у дітей з 2 років і старше. Повільний розвиток хвороби, але прогресуюче. Направлена на пригнічення фізичної активності. Спочатку дитина рухається, ходить, піднімається і спускається по сходах, а згодом рухова активність викликає труднощі. Це призводить так само спінальної-м’язової атрофії. В першу чергу страждає м’язова система ніг, пізніше спини, шиї і в кінці верхнього плечового пояса. Самоуход можливий досить тривалий час, але в будь-якому випадку цей тип так само веде до інвалідності.
Тип 4. Доросла форма захворювання
 Зустрічається рідко і починає свій розвиток з 30 років життя людини. В основному задіяні шийні і головні м’язи, пацієнт може повністю себе обслуговувати. Ознаками спінальної-м’язової атрофії може бути слабка мімічна діяльність м’язів обличчя, сіпання мови, деяке обмеження рухливості голови. Як правило, це вважається успішним результатом захворювання.
Зустрічається рідко і починає свій розвиток з 30 років життя людини. В основному задіяні шийні і головні м’язи, пацієнт може повністю себе обслуговувати. Ознаками спінальної-м’язової атрофії може бути слабка мімічна діяльність м’язів обличчя, сіпання мови, деяке обмеження рухливості голови. Як правило, це вважається успішним результатом захворювання.
Діагностика
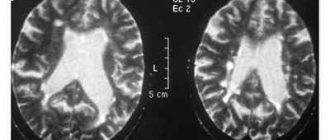
Діагностують за сукупністю перших ознак захворювання. Для підтвердження діагнозу у пацієнта беруть біопсію потенційно ураженого м’яза, лабораторними методами уточнюють патологічні зміни. Для того, щоб виявити патологію у дитини або дорослого призначають МРТ і КТ. Аналіз ТМСВ широко використовується в діагностиці захворювання, в при генетичному скринінгу потенційно підозрюваних хворих. ТМС — транскраніальна магнітна стимуляція, діагностичний амбулаторний метод, заснований на стимуляції кори головного мозку за допомогою коротких магнітних імпульсів, не приносить будь-яких хворобливих відчуттів. Профілактика полягає в бесіді з лікарем-генетиком пари, у якій це захворювання вже є в сімейному анамнезі або є ген СМА. Складається генеалогічне древо і робиться висновок про процентне співвідношення і ризик передачі спадкового захворювання дитині. Так само проводиться біопсія ворсин хоріона. Але жоден метод діагностики або профілактики не дає 100% позитивний результат про наявність генетичного захворювання у дітей.
Лікування
 Медицина ще не винайшла засіб лікування спінальної-м’язової атрофії. Вся суть лікування спрямована на підтримку життєдіяльності хворого і з метою уникнення будь-яких ускладнень. Що входить в сукупність заходів з лікування захворювання:
Медицина ще не винайшла засіб лікування спінальної-м’язової атрофії. Вся суть лікування спрямована на підтримку життєдіяльності хворого і з метою уникнення будь-яких ускладнень. Що входить в сукупність заходів з лікування захворювання:
- Лікарська терапія, при якій нервовий імпульс буде краще проходити від центрального відділу нервової системи до периферичних відділах. Так само лікарська терапія повинна включати препарати з вмістом калію.
- Вітамінотерапія, обов’язково комплексна. Групи В.
- Препарати ноотропной терапії, для відновлення кровопостачання в нервовій тканині.
- Фізіологічні процедури, як закріплення названої вище терапії, наприклад, парафінові ванни.
- Обов’язковою є масаж при спінальної м’язової атрофії, для м’язового тонусу.
- Для зміцнення зв’язок використовується лікувально — профілактична фізкультура.
У найважчих формах, потрібно реанімаційна заходи. Багато вчених і медики працюють над таким препаратом, який би заповнював певний білок при цьому захворюванні. Це дає надію на повне одужання від тяжкої генетичної хвороби, нехай навіть штучним шляхом і довічним вживанням лікарських засобів.