Хвороба Крейтцфельдта-Якоба (ХКЯ) або як її ще називають синдром кортико-стріоспінальной дегенерації ставитися до патологій з швидкопрогресуючим плином. Для неї властиві дистрофічні зміни тканин головного мозку. Таке явище зазвичай веде до розвитку деменції (недоумства) і швидкого летального результату. Симптоматика патології фактично ідентична Альцгеймера проявам. Однак, на відміну від них, хвороба Крейтцфельдта-Якоба значно швидше розвивається. Такий вид патології може бути класичним, тобто викликатися спонтанно або бути наслідком генних мутацій і набутим. До другого типу відносяться зараження від хворої тварини або людини, а також вживання інфікованої яловичини. Останній випадок був виявлений в 1986 році після спалаху епідемії коров’ячого сказу (нейродегенератівнойВ пріонів хвороби) в Англії. З тих пір хвороба Крейтцфельдта-Якоба вважається одним із проявів пріонів захворювання. Вона також називається губчастим енцефалітом через зовнішнього вигляду змінених тканин головного мозку. Виникають такі порушення через вплив пріонів, які є інфекційними агентами. Губчастий енцефаліт проявляється вкрай рідко як в класичному спонтанному варіанті, так і внаслідок коров’ячого сказу. За статистикою щороку лише в 1 випадку на 1 мільйон чоловік населення ставиться такий діагноз і в основному у вікових людей. 
Причини
Губкообразная енцефалопатія, що викликається хворобою, є наслідком впливу патогенних білків (протеїнів), які називаються пріонами. У нормі вони абсолютно нешкідливі, але внаслідок впливу певних факторів протеїни піддаються змінам. Зрозуміти, як виникає синдром Крейтцфельдта-Якоба можна, поглянувши на її види:
- Спонтанний прояв. Такий тип виникнення синдрому кортико-стріоспінальной дегенерації відноситься до класичного і не має явних причин появи;
- Генетична аномалія. В Америці Губкообразная енцефалопатія в 10% випадків виникає через генної мутації. Такий різновид називається сімейною хворобою Крейтцфельдта-Якоба і передається переважно у спадок;
- Зараження. У рідкісних випадках синдром кортико-стріоспінальной дегенерації проявляється у людей після попадання в кров інфікованих частинок, отриманих від хворої людини або тварини. Таке явище можливе після пересадок шкірного покриву або інших тканин. Іноді зараження відбувається внаслідок оперативного втручання на головному мозку. У цьому випадку передача йде з поверхні інструментів, так як не можна вбити пріони звичайними методами стерилізації. Заразитися від тваринного також можливо, наприклад, через прийом лікарських засобів, отриманих з його головного мозку. У цьому випадку мова йде про синдром ятрогенного типу;
- Коров’ячий сказ. Випадки поширення такого різновиду хвороби вкрай рідкісні. Підхопити її можна, вживаючи погано приготовану яловичину або працюючи з сирим інфікованим м’ясом. Цей вид патології називається коров’яча губчаста енцефалопатія.
Групи ризику
Більшість випадків синдрому кортико-стріоспінальной дегенерації є спонтанними і встановити причини їх розвитку не представляється можливим. Однак для цього патологічного процесу властиві певні чинники, що впливають на його розвиток, а саме:
- Вік хворого. Зазвичай до 60-65 років виникає спонтанний тип хвороби, а від сімейної різновиди люди страждають після 45-50 років. Зараження хворобою Крейтцфельдта-Якоба і зовсім не має вікових рамок. Для прикладу під час великої епідемії коров’ячого сказу в Англії більшості хворих було менше 35-40 років;
- Генетична схильність. Такий фактор зазвичай стосується сімейної різновиди синдрому кортико-стріоспінальной дегенерації. Виникає він внаслідок генної мутації і передається по аутосомно-домінантним типом. Такий механізм поширення означає те, що досить наявності генного дефекту у одного з батьків і при цьому буде близько 50% ймовірності народження дитини з цією аномалією. Однак спадкова схильність також впливає і на інші форми патології. У хворих з ятрогенним ХКЯ і коров’ячої губчастої енцефалопатією найчастіше є дефектні гени. Таке явище свідчить про те, що генетична схильність збільшує шанси заразитися пріонів патологією від хворої людини або тварини;
- Вплив інфікованого об’єкта. Пересадка тканин від інфікованої тварини або людини, яка страждає від ХКЯ, здатна впливати на розвиток патологічного процесу. Особливо якщо у хворого є генні аномалії.

Сьогодні підхопити коров’ячий сказ вкрай складно, так як її спалахи виникають рідко і переважно в слаборозвинених країнах. Для прикладу заразитися цією патологією в Англії майже нереально. Пов’язано це з ретельним контролем і лише в 1 блюді на 10 мільярдів можна зустріти заражену яловичину.
Симптоми
Розвивається ХКЯ блискавично і явні ознаки деменції стають помітні вже через кілька місяців. Серед її базових проявів можна виділити наступні:
- Спонтанні і швидкі рухи;
- Проблеми з ковтанням;
- Зміна поведінки;
- Почуття тривоги;
- Депресійне стан;
- Погіршення пам’яті;
- Зниження розумових здібностей;
- Погіршення зору;
- Порушення ритму сну;
- Виникнення мовних дефектів.
Для хвороби Крейтцфельдта-Якоба властива швидка прогресування, тому в лічені місяці стан хворого значно погіршується. Більшість людей впадає в коматозний стан. Летальний результат часто трапляється через дихальної або серцевої недостатності, а також внаслідок інфекцій, здатних викликати пневмонію та інші схожі патології. Прогноз вкрай невтішний.
Якщо патологічний процес є наслідком вживання інфікованої яловичини, то симптоми розладу психіки проявляються не так очевидно. Порушення когнітивних функцій і зовсім виникає на більш пізніх термінах. Однак результат цього різновиду хвороби аналогічний класичному типу ХКЯ. Люди вмирають приблизно через 2 роки від початку розвитку патологічного процесу.
Діагностика

Після виявлення перших ознак захворювання, необхідно негайно відправитися в лікарню. Дізнатися про те, чи є синдром кортико-стріоспінальной дегенерації у людини чи ні можна, але лише за допомогою взяття на аналіз тканини головного мозку (біопсії) або посмертно. У більшості випадків лікаря залишається лише припускати наявність патології у пацієнта. Для цього він дивиться на сімейний анамнез, що виникає симптоматику і на результати інструментального дослідження:
- Електроенцефалографія. Її суть полягає в вимірі електричної мозкової активності;
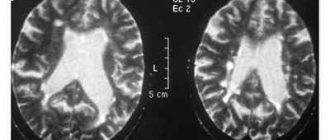
- Магнітно-резонансна томографія. З її допомогою лікар може детально розглянути сіра і біла мозкова речовина і виявити патологічні відхилення, в тому числі і дистрофічного характеру;
- Люмбальна пункція. Таку процедуру проводять для вилучення на аналіз ліквору (спинномозкової рідини);
- Біопсія мигдалин. За її складу лікар може судити про наявність в організмі інфекції, що викликає коров’ячий сказ, але такий метод діагностики менш точний, ніж інші.
За результатами обстеження невропатолог може виявити порушення властиві хвороби Крейтцфельдта-Якоба. Однак точно дізнатися про наявність патогенних пріонів в організмі можна лише за допомогою біопсії тканин головного мозку.
Курс терапії
На сьогоднішній день так і не було придумано жодного чинного ліки проти ХКЯ. За кілька десятків років вчені не змогли зрушити в цьому питанні, і будь-яка форма такої патології з часом призводить до летального результату. Єдине чим може допомогти сучасна медицина, так це полегшити загальний стан хворого за допомогою симптоматичної терапії.
Ускладнення
Синдром кортико-стріоспінальной дегенерації може привести до смерті за півроку, тому вважається вкрай небезпечним. Хворі вже в перші місяці починають вести себе дивно, відштовхують близьких людей і навіть перестають їх впізнавати. Поступово вони забувають про те, що потрібно митися, спати, їсти і т. Д. 
Серед фізичних ускладнень, які можуть призвести до летального результату можна виділити наступні:
- Серцева і дихальна недостатність;
- Розвиток інфекційних захворювань.
Хвороба Крейтцфельдта-Якоба є смертельним вироком, від якого немає лікування. Однак вчені продовжують вести дослідження, мета яких зупинити розвиток патології, тому хворим не потрібно впадати у відчай і намагатися продовжувати своє життя і далі. В такому випадку завжди залишається шанс дочекатися бажаного ліки.